理论计算入门手册 从分子模拟到量子计算的全景概览
理论计算作为现代科学研究与工业应用的重要工具,已渗透到材料科学、药物设计、能源催化等多个前沿领域。本文旨在为初学者提供一份清晰、系统的概念指南,涵盖从经典方法到前沿技术的核心要点。
一、分子模拟:经典体系的动态洞察
分子模拟主要基于牛顿力学,通过数值方法模拟原子与分子的运动轨迹,从而揭示体系的宏观性质与动态过程。
- 核心方法:分子动力学(MD)与蒙特卡洛(MC)方法是两大支柱。MD通过求解运动方程跟踪粒子随时间的演化,适用于研究扩散、相变等动态行为;MC则基于概率抽样探索构型空间,常用于计算热力学平衡性质。
- 力场关键:模拟精度高度依赖于力场(如AMBER、CHARMM),其通过经验参数描述原子间的相互作用(键结、非键结等),在生物大分子、聚合物体系中应用广泛。
- 应用场景:蛋白质折叠、药物-受体结合、材料力学性能预测等。
二、量子化学:电子结构的精确描绘
量子化学从量子力学出发,直接处理电子结构问题,适用于化学键形成、反应机理等需要电子层次信息的场景。
- 从头算方法:不依赖经验参数,仅基于物理常数求解薛定谔方程。哈特里-福克(HF)方法是起点,但忽略电子关联效应;后哈特里-福克方法(如MP2、CCSD)通过微扰或组态相互作用提升精度。
- 密度泛函理论(DFT):平衡精度与效率的标杆。通过电子密度而非波函数描述体系,交换关联泛函(如B3LYP、PBE)的选择至关重要。DFT广泛用于催化反应、光谱预测及材料电子性质计算。
- 基组选择:基组(如6-31G*、def2-TZVP)是描述原子轨道的数学函数集,其大小与质量直接影响计算成本与结果可靠性。
三、第一性原理计算:材料设计的基石
第一性原理常与DFT等同,强调“从第一性原理出发”无需实验输入,是材料模拟的核心工具。
- 核心优势:能够预测未知材料的性质(如能带结构、磁学特性),指导新型功能材料(拓扑绝缘体、钙钛矿太阳能电池等)的设计。
- 典型软件:VASP、Quantum ESPRESSO、ABINIT等开源或商业软件实现了高效并行计算,支持周期性边界条件,完美适配晶体、表面等扩展体系。
- 多尺度桥接:第一性原理结果可为分子模拟提供高精度参数,实现从电子到宏观的跨尺度建模。
四、有限元分析:连续介质的多物理场仿真
有限元法(FEM)将连续体离散为有限个单元,通过数值求解偏微分方程模拟结构、流体、热传导等物理过程。
- 工程利器:在机械工程、航空航天、土木建筑等领域用于应力分析、振动模态、热分布计算等,软件如ANSYS、COMSOL已成为行业标准。
- 与原子模拟的互补:有限元处理宏观连续介质,而分子/量子方法描述微观离散原子,两者结合可实现从原子缺陷到工程构件的一体化设计。
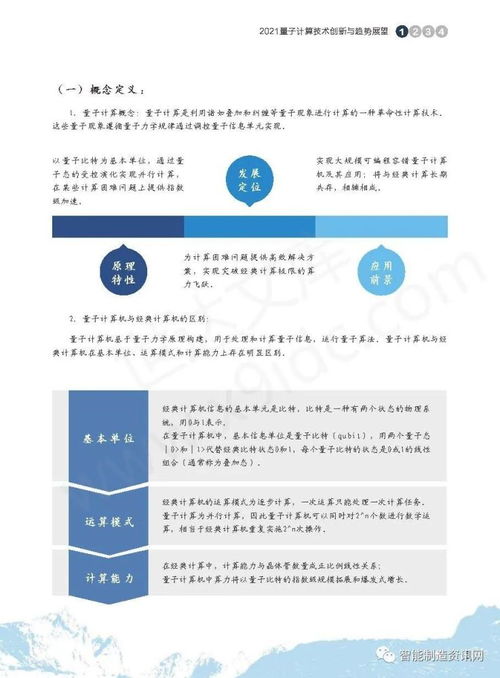
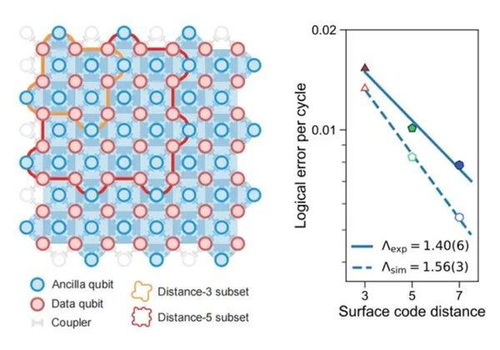
五、量子计算技术服务:下一代计算范式
量子计算利用量子比特的叠加与纠缠,有望在特定问题上指数级超越经典计算机,正逐步从理论走向实用化服务。
- 算法突破:量子化学是量子计算的重要应用场景。VQE(变分量子本征求解器)等算法可在量子硬件上模拟分子电子结构,为药物设计、催化剂开发提供新途径。
- 云平台接入:IBM Q、Amazon Braket等云服务已提供量子计算资源,允许研究者远程运行量子线路,降低入门门槛。
- 混合计算模式:近期以实现“量子优势”为目标,经典-量子混合算法(如量子机器学习)成为当前技术服务的焦点,助力优化、材料发现等复杂任务。
方法选择与融合趋势
理论计算方法各具特色:分子模拟擅长大规模体系与动态过程;量子化学与第一性原理精于电子结构;有限元主宰宏观工程仿真;量子计算则代表未来突破方向。实际研究中,多尺度、多物理场耦合已成为常态(如QM/MM将量子区与经典区结合),而云计算与人工智能的融入正加速计算范式的革新。初学者可根据体系尺寸、精度需求与计算资源,选择合适工具,并保持对交叉技术发展的关注,以充分利用理论计算揭示自然奥秘、驱动工业创新的强大潜力。
如若转载,请注明出处:http://www.ghdsfdszs.com/product/71.html
更新时间:2026-06-19 08:14:08